Spatial transcriptome-Stereo-seq
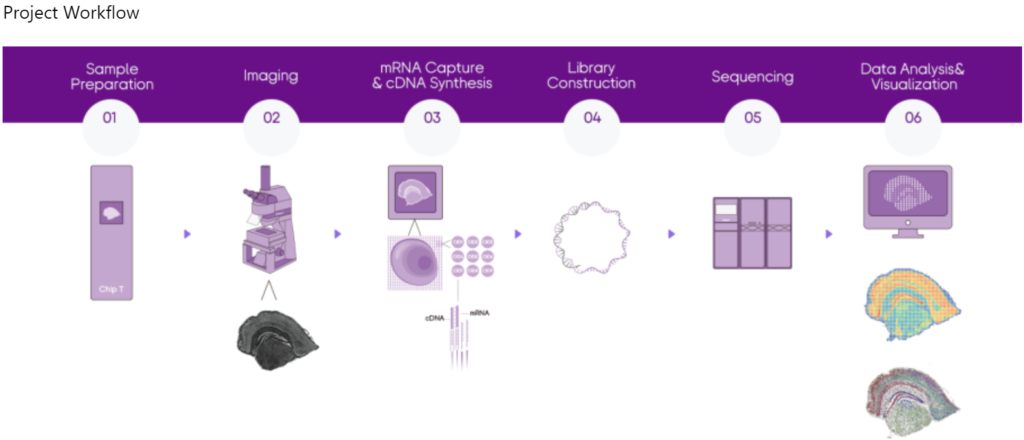
Stereo-seq, a spatio-temporal omics technology independently developed by BGI Genomics, captures mRNA from tissue section by stereo chips and restores the spatial context by utilizing the spatial barcode (Coordinate ID, CID), thus establishing a solid research foundation for further understanding the relationship between gene expression, morphology of cells and local environment.
Stereo-seq is a pioneering tool that achieves Nanoscale Resolution: can theoretically achieve a 100% cell capture rate, obtaining more informative and accurate cell clustering results.
Stereo-seq provides centimeter-scale panoramic field of view, a maximum field of view of 13 cm x 13 cm, enabling the rendering of a panoramic molecular cell map of organs and life. Stereo-seq recognizes the location of the nucleus through fluorescent imaging, and in combination with the algorithm, the expression map can be achieved at approximate single-cell level.